
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
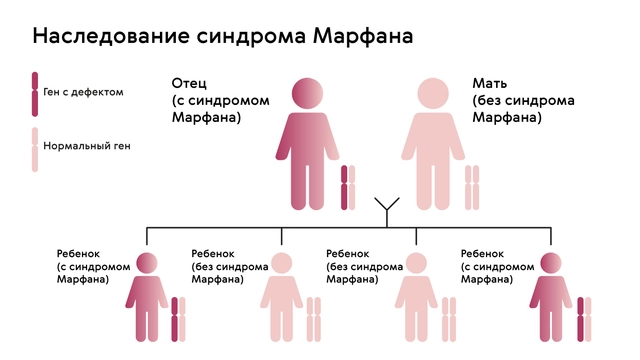
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
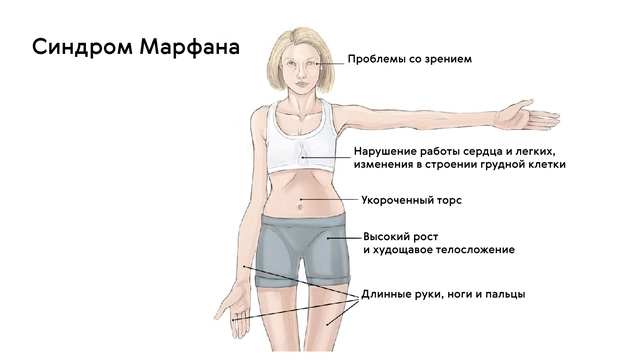
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
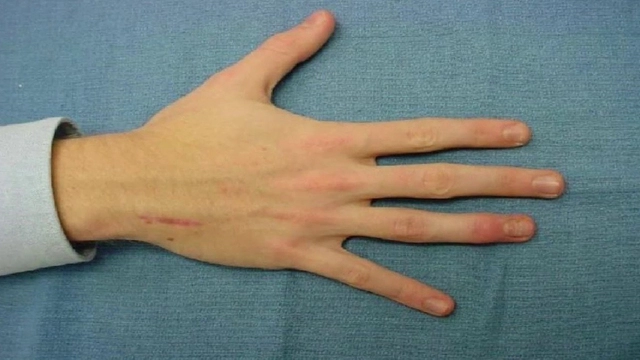
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
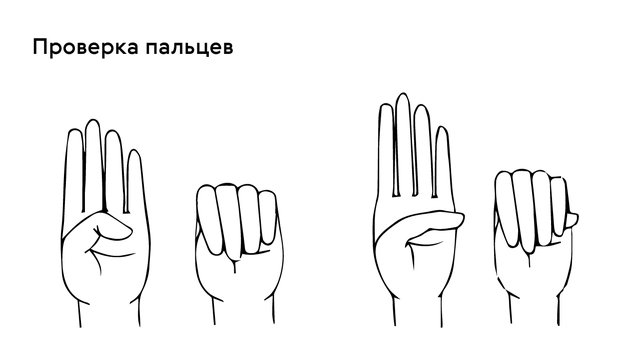
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
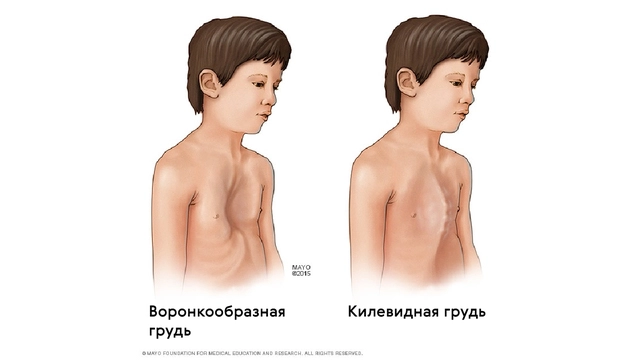
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Кожа
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).

Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Источники
- Отделение синдрома Марфана. Андалузский институт высокотехнологичной кардиологии (Бенальмадена и Фуэнхирола, Малага, Испания).
- Dadkhah, E., Ziaee, M., Davari, M. H., Kazemi, T., & Abbaszadegan, M. R. (2012). Informative STR Markers for Marfan Syndrome in Birjand, Iran. Iranian journal of basic medical sciences, 15(5), 1020–1025.
- Прийма, Н. Ф., Попов, В. В., Комолкин, И. А., & Афанасьев, А. П. (2013). Аневризма аорты у пациента с синдромом Марфана. Педиатр, 4 (1), 100-108.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. doi: 10.1136/jmg.2009.072785. PMID: 20591885.













